![]() Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
1. DÉNOMINATION DU MÉDICAMENT
EXJADE 90 mg comprimés pelliculés
EXJADE 180 mg comprimés pelliculés
EXJADE 360 mg comprimés pelliculés
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
EXJADE 90 mg comprimés pelliculés
Chaque comprimé pelliculé contient 90 mg de déférasirox.
EXJADE 180 mg comprimés pelliculés
Chaque comprimé pelliculé contient 180 mg de déférasirox.
EXJADE 360 mg comprimés pelliculés
Chaque comprimé pelliculé contient 360 mg de déférasirox.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé.
EXJADE 90 mg comprimés pelliculés
Comprimé pelliculé bleu clair, ovoïde, biconvexe, avec des bords biseautés et gravés (NVR sur une face et 90 sur l’autre). Dimensions approximatives du comprimé de 10,7 mm x 4,2 mm.
EXJADE 180 mg comprimés pelliculés
Comprimé pelliculé bleu, ovoïde, biconvexe, avec des bords biseautés et gravés (NVR sur une face et 180 sur l’autre). Dimensions approximatives du comprimé de 14 mm x 5,5 mm.
EXJADE 360 mg comprimés pelliculés
Comprimé pelliculé bleu foncé, ovoïde, biconvexe, avec des bords biseautés et gravés (NVR sur une face et 360 sur l’autre). Dimensions approximatives du comprimé de 17 mm x 6,7 mm.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
EXJADE est indiqué dans le traitement de la surcharge en fer chronique secondaire à des transfusions sanguines fréquentes (7 ml/kg/mois de concentrés érythrocytaires) chez les patients de 6 ans et plus qui présentent une bétâ-thalassémie majeure.
EXJADE est aussi indiqué dans le traitement de la surcharge en fer chronique secondaire à des transfusions sanguines lorsque le traitement par la déféroxamine est contre-indiqué ou inadapté chez les groupes de patients suivants :
- les patients pédiatriques âgés de 2 à 5 ans présentant une bétâ-thalassémie majeure avec une surcharge en fer chronique secondaire à des transfusions sanguines fréquentes (≥7 ml/kg/mois de concentrés érythrocytaires).
- les patients adultes et pédiatriques âgés de 2 ans et plus présentant une bétâ-thalassémie majeure avec une surcharge en fer chronique secondaire à des transfusions sanguines peu fréquentes (<7 ml/kg/mois de concentrés érythrocytaires).
- Les patients adultes et pédiatriques âgés de 2 ans et plus présentant d’autres types d’anémies.
EXJADE est également indiqué dans le traitement de la surcharge en fer chronique nécessitant un traitement chélateur du fer chez les patients de 10 ans et plus présentant des syndromes thalassémiques non dépendants des transfusions, lorsque le traitement par déféroxamine est contre-indiqué ou inadapté.
4.2 Posologie et mode d’administration
Le traitement par EXJADE doit être initié et conduit par des médecins expérimentés dans le traitement de la surcharge en fer.
Posologie
Surcharge en fer post-transfusionnelle
Il est recommandé que le traitement soit initié après la transfusion d’environ 20 unités de concentrés érythrocytaires (CE) (soit 100 ml/kg) ou lorsque le suivi clinique met en évidence la présence d’une surcharge en fer (par exemple ferritinémie >1 000 µg/l). Les doses (en mg/kg) doivent être calculées et arrondies au comprimé le plus proche.
Les objectifs du traitement chélateur du fer sont d’éliminer le fer apporté par les transfusions et si nécessaire de réduire la surcharge en fer existante.
Une attention particulière doit être appliquée pendant le traitement chélateur du fer pour minimiser le risque de chélation excessive chez tous les patients traités (voir rubrique 4.4).
Dans l’Union Européenne, les médicaments contenant du déférasirox sont disponibles sous forme de comprimés pelliculés et de comprimés dispersibles commercialisés sous différents noms de spécialités en tant qu’alternatives génériques d’EXJADE. En raison des différents profils pharmacocinétiques, une réduction de 30% de la dose de la forme comprimés pelliculés d’EXJADE est nécessaire par rapport à la dose recommandée pour la forme comprimés dispersibles d’EXJADE (voir rubrique 5.1).
Tableau 1 Doses recommandées en cas de surcharge en fer post-transfusionnelle
| Comprimés pelliculés | Transfusions |
| Ferritinémie |
Dose initiale | 14 mg/kg/jour | Après 20 unités de concentrés érythrocytaires (environ 100 ml/kg) | ou | >1 000 µg/l |
Dose initiale alternative | 21 mg/kg/jour | >14 ml/kg/mois de concentrés érythrocytaires (environ >4 unités/mois pour un adulte) |
|
|
| 7 mg/kg/jour | <7 ml/kg/mois de concentrés érythrocytaires (environ <2 unités/mois pour un adulte) |
|
|
Chez les patients bien équilibrés avec un traitement par la déféroxamine | Un tiers de la dose de déféroxamine |
|
|
|
Surveillance |
|
|
| Mensuelle |
Taux ciblé |
|
|
| 500‑1 000 µg/l |
|
|
|
|
|
Paliers d’ajustement | Augmentation |
|
| >2 500 µg/l |
3,5 ‑ 7 mg/kg/jour |
|
|
| |
Diminution |
|
|
| |
3,5 ‑ 7 mg/kg/jour |
|
| ≤2 500 µg/l | |
Chez les patients traités à des doses >21 mg/kg/jour |
|
|
| |
|
|
| 500‑1 000 µg/l | |
Dose maximale | 28 mg/kg/jour |
|
|
|
Envisager l’interruption du traitement |
|
|
| <500 µg/l |
Dose initiale
La dose journalière initiale recommandée d’EXJADE comprimés pelliculés est de 14 mg/kg de poids corporel.
Une dose initiale journalière de 21 mg/kg d’EXJADE comprimés pelliculés peut être envisagée pour les patients qui nécessitent une réduction de leur surcharge en fer et qui reçoivent plus de 14 ml/kg/mois de concentrés érythrocytaires (approximativement >4 unités/mois pour un adulte).
Une dose initiale journalière de 7 mg/kg d’EXJADE comprimés pelliculés peut être envisagée pour les patients qui ne nécessitent pas une réduction de leur surcharge en fer et qui reçoivent moins de 7 ml/kg/mois de concentrés érythrocytaires (approximativement <2 unités/mois pour un adulte). La réponse du patient doit être contrôlée et une augmentation de la dose devra être envisagée si une efficacité suffisante n’est pas obtenue (voir rubrique 5.1).
Chez les patients déjà bien équilibrés avec un traitement par la déféroxamine, une dose initiale d’EXJADE comprimés pelliculés correspondant numériquement au tiers de celle de la déféroxamine pourra être envisagée (par exemple le traitement d’un patient par 40 mg/kg/jour de déféroxamine pendant 5 jours par semaine (ou équivalent) pourra être substitué par une dose initiale d’EXJADE comprimés pelliculés de 14 mg/kg/j). Lorsque la dose journalière résultante est de moins de 14 mg/kg/j, la réponse du patient devra être contrôlée et une augmentation de la dose devra être envisagée si une efficacité suffisante n’a pas été obtenue (voir rubrique 5.1).
Ajustement de la dose
Il est recommandé de contrôler la ferritinémie tous les mois et d’ajuster la dose d’EXJADE comprimés pelliculés si nécessaire, tous les 3 à 6 mois en fonction des variations de la ferritinémie. Les ajustements de la dose doivent être réalisés par paliers de 3,5 à 7 mg/kg et doivent être adaptés à la réponse individuelle du patient et aux objectifs thérapeutiques (maintien ou réduction de la surcharge en fer). Chez les patients qui ne sont pas correctement contrôlés à des doses de déférasirox comprimés pelliculés de 21 mg/kg (par exemple ferritinémie persistante au-dessus de 2500 µg/l et n’ayant pas tendance à diminuer avec le temps), des doses allant jusqu’à 28 mg/kg peuvent être envisagées. Les données d’efficacité et de sécurité d’emploi à long terme provenant d'études cliniques menées avec EXJADE comprimés dispersibles à des doses supérieures à 30 mg/kg sont actuellement limitées (264 patients suivis pendant un 1 an en moyenne après augmentation de dose). Si seulement un très faible contrôle de l’hémosidérose à des doses de déférasirox comprimés pelliculés supérieures à 21 mg/kg est atteint (dose de comprimés pelliculés équivalente à 30 mg/kg de comprimés dispersibles), une autre augmentation (jusqu’à un maximum de 28 mg/kg) pourrait ne pas entraîner un contrôle satisfaisant et d’autres options thérapeutiques devront donc être envisagées. Si un contrôle satisfaisant n’est pas atteint à des doses supérieures à 21 mg/kg, un traitement à de telles doses ne devra pas être maintenu et d’autres options thérapeutiques devront être envisagées quand cela sera possible. Les doses supérieures à 28 mg/kg sont déconseillées car l’expérience avec des doses supérieures à cette valeur est limitée (voir rubrique 5.1).
Chez les patients traités à des doses supérieures à 21 mg/kg, des réductions de dose par paliers de 3,5 à 7 mg/kg devront être envisagées quand le contrôle a été atteint (par exemple, ferritinémie ≤2500 µg/l de façon persistante et ayant tendance à diminuer avec le temps). Chez les patients pour lesquels la ferritinémie a atteint son objectif (habituellement entre 500 et 1000 µg/l), des réductions de dose par paliers de 3,5 à 7 mg/kg devront être envisagées afin de maintenir la ferritinémie dans ces valeurs cibles et de minimiser le risque de chélation excessive. Si la ferritinémie chute de façon persistante en dessous de 500 µg/l, l’interruption du traitement doit être envisagée (voir rubrique 4.4).
Syndromes thalassémiques non dépendants des transfusions
Le traitement chélateur du fer ne doit être initié que lorsque la présence d'une surcharge en fer a été mise en évidence (concentration hépatique en fer [CHF] ≥5 mg Fe/g de poids sec ou ferritinémie persistante >800 µg/l). La mesure de la CHF est la méthode de choix pour évaluer la surcharge en fer, elle doit être employée lorsqu’elle est disponible. Une attention particulière doit être appliquée pendant le traitement chélateur afin de minimiser le risque de chélation excessive chez tous les patients traités (voir rubrique 4.4).
Dans l’Union Européenne, les médicaments contenant du déférasirox sont disponibles sous forme de comprimés pelliculés et de comprimés dispersibles commercialisés sous différents noms de spécialités en tant qu’alternatives génériques d’EXJADE. En raison des différents profils pharmacocinétiques, une réduction de 30% de la dose de la forme comprimés pelliculés d’EXJADE est nécessaire par rapport à la dose recommandée pour la forme comprimés dispersibles d’EXJADE (voir rubrique 5.1).
Tableau 2 Doses recommandées en cas de syndromes thalassémiques non dépendants des transfusions
| Comprimés pelliculés | Concentration hépatique en fer (CHF)* |
| Ferritinémie |
Dose initiale | 7 mg/kg/jour | ≥5 mg Fe/g de poids sec | ou | >800 µg/l |
Surveillance |
|
|
| Mensuelle |
Paliers d’ajustement | Augmentation | ≥7 mg Fe/g de poids sec | ou | >2 000 µg/l |
3,5 ‑ 7 mg/kg/jour |
|
|
| |
Diminution | <7 mg Fe/g de poids sec | ou | ≤2 000 µg/l | |
3,5 ‑ 7 mg/kg/jour |
|
|
| |
Dose maximale | 14 mg/kg/jour |
|
|
|
| 7 mg/kg/jour |
|
|
|
| 7 mg/kg/jour | Non évalué | et | ≤2 000 µg/l |
Interruption |
| <3 mg Fe/g de poids sec | ou | <300 µg/l |
Reprise du traitement |
| Non recommandée | ||
*La CHF est la méthode privilégiée pour la détermination des surcharges en fer.
Dose initiale
La dose journalière initiale recommandée d'EXJADE comprimés pelliculés chez les patients présentant des syndromes thalassémiques non dépendants des transfusions est de 7 mg/kg de poids corporel.
Ajustement de la dose
Il est recommandé de contrôler la ferritinémie tous les mois afin d’évaluer la réponse du patient au traitement et de minimiser le risque de chélation excessive (voir rubrique 4.4). Après 3 à 6 mois de traitement, une augmentation de la dose du déférasirox comprimés pelliculés par paliers de 3,5 à 7 mg/kg doit être envisagée si la CHF du patient est ≥7 mg Fe/g de poids sec ou si la ferritinémie est >2000 µg/l de façon persistante et n'a pas tendance à diminuer et si le patient tolère bien le médicament. Les doses d’EXJADE comprimés pelliculés supérieures à 14 mg/kg ne sont pas recommandées car il n'y a pas d'expérience à ces doses chez les patients qui présentent des syndromes thalassémiques non dépendants des transfusions.
Chez les patients pédiatriques et adultes pour qui la CHF n'a pas été évaluée et pour qui la ferritinémie est ≤2000 µg/l, la dose d’EXJADE comprimés pelliculés ne doit pas dépasser 7 mg/kg.
Chez les patients pour qui la dose a été augmentée à plus de 7 mg/kg, une réduction de la dose à 7 mg/kg ou moins est conseillée lorsque la CHF est <7 mg Fe/g de poids sec ou lorsque la ferritinémie est ≤2000 µg/l.
Arrêt du traitement
Lorsqu'une charge corporelle en fer satisfaisante a été obtenue (CHF <3 mg Fe/g de poids sec ou ferritinémie <300 µg/l), le traitement doit être arrêté. Il n’y a pas de données disponibles sur la reprise du traitement chez des patients ayant de nouveau accumulé du fer après avoir atteint une charge corporelle en fer satisfaisante, par conséquent la reprise du traitement ne peut être recommandée.
Populations particulières
Sujets âgés (≥ à 65 ans)
Les recommandations sur la posologie chez le sujet âgé sont les mêmes que celles décrites ci-dessus. Dans les études cliniques, la fréquence des effets indésirables a été plus élevée chez les sujets âgés que chez les sujets plus jeunes (en particulier, les diarrhées). Une surveillance attentive des effets indésirables nécessitant un ajustement de la dose est nécessaire chez ces patients.
Population pédiatrique
Surcharge en fer post-transfusionnelle :
Les recommandations sur la posologie chez l’enfant âgé de 2 à 17 ans présentant une surcharge en fer post transfusionnelle sont les mêmes que chez l’adulte (voir rubrique 4.2). Il est recommandé de contrôler la ferritinémie tous les mois afin d’évaluer la réponse du patient au traitement et de minimiser le risque de chélation excessive (voir rubrique 4.4). L’évolution du poids au cours du temps chez l’enfant doit être prise en considération dans le calcul de la dose.
Chez les enfants âgés de 2 à 5 ans présentant une surcharge en fer post transfusionnelle, l’exposition est plus faible que chez l’adulte (voir rubrique 5.2). Ce groupe d’âge peut nécessiter des doses plus élevées que celles nécessaires chez l’adulte. Toutefois, la dose initiale devra être la même que celle de l’adulte, elle sera adaptée ensuite individuellement.
Syndromes thalassémiques non dépendants des transfusions :
Chez les enfants présentant un syndrome thalassémique non dépendant des transfusions, la dose d’EXJADE comprimés pelliculés ne doit pas dépasser 7 mg/kg. Chez ces patients, un contrôle plus étroit de la CHF et de la ferritinémie est indispensable pour éviter une chélation excessive (voir rubrique 4.4). En plus des contrôles mensuels de la ferritinémie, la CHF doit être contrôlée tous les trois mois lorsque la ferritine sérique est ≤800 µg/l.
Enfants âgés de 0 à 23 mois :
La sécurité et l’efficacité d’EXJADE chez les enfants âgés de 0 à 23 mois n’ont pas été établies. Aucune donnée n’est disponible.
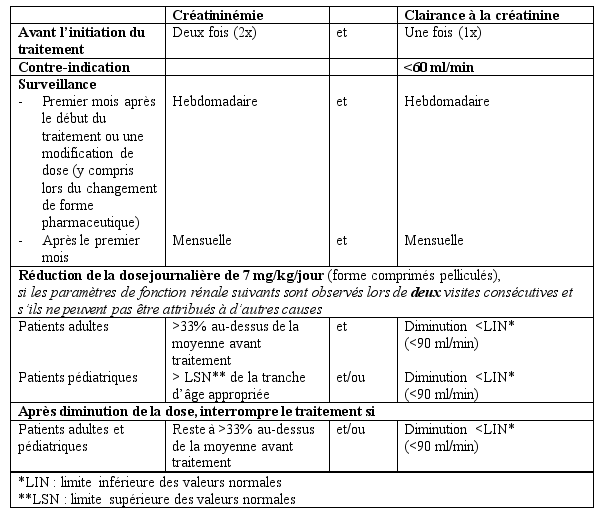
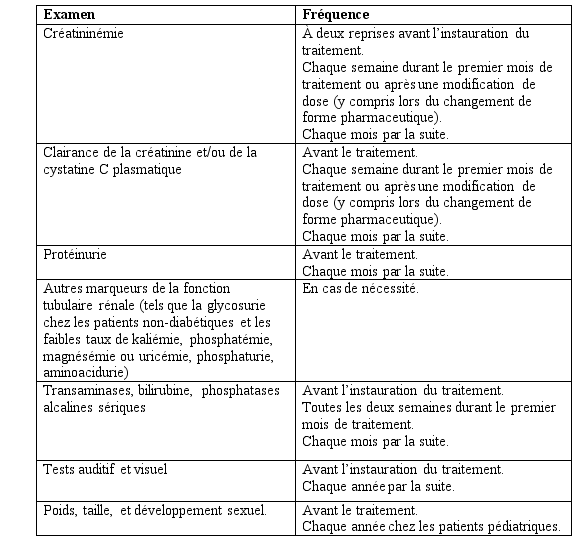
Patients avec insuffisance rénale
EXJADE n’a pas été étudié chez les patients ayant une insuffisance rénale et il est contre-indiqué chez les patients présentant une clairance de la créatinine estimée inférieure à 60 ml/min (voir rubrique 4.3 et 4.4).
Patients avec insuffisance hépatique
EXJADE n’est pas recommandé chez les patients ayant une insuffisance hépatique sévère (Child-Pugh Classe C). Chez les patients atteints d’une insuffisance hépatique modérée (Child-Pugh Classe B), la dose doit être considérablement réduite suivie d’une augmentation progressive sans dépasser 50% de la dose de traitement recommandée pour les patients ayant une fonction hépatique normale (voir rubriques 4.4 et 5.2) et EXJADE doit être utilisé avec précautions chez ces patients. La fonction hépatique devra être contrôlée chez tous les patients avant traitement, toutes les 2 semaines pendant le premier mois, puis tous les mois (voir rubrique 4.4).
Mode d’administration
Par voie orale.
Les comprimés pelliculés doivent être avalés entiers avec de l’eau. Chez les patients qui ne peuvent pas avaler les comprimés en entier, les comprimés pelliculés peuvent être écrasés et administrés en saupoudrant la dose totale dans une petite quantité de nourriture non solide, tel qu’un yaourt ou une compote de pomme par exemple. La dose doit être prise immédiatement et complètement et ne pas être conservée pour une utilisation ultérieure.
Les comprimés pelliculés doivent être pris une fois par jour, de préférence à la même heure tous les jours, et peuvent être pris à jeun ou avec un repas léger (voir rubriques 4.5 et 5.2).
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
L’association à d’autres traitements chélateurs du fer car la tolérance de ces associations n’a pas été étudiée (voir rubrique 4.5).
Patients présentant une clairance de la créatinine estimée inférieure à 60 ml/min.
4.8 Effets indésirables
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés dans les études cliniques, au cours du traitement chronique par déférasirox comprimés dispersibles chez des patients adultes et pédiatriques comprennent les troubles gastro-intestinaux (principalement nausées, vomissements, diarrhées, ou douleurs abdominales) et les éruptions cutanées. Les diarrhées sont rapportées plus fréquemment chez les enfants âgés de 2 à 5 ans que chez les personnes âgées. Ces réactions sont doses-dépendantes, essentiellement légères à modérées, généralement transitoires et pour la plupart résolutives même si le traitement est poursuivi.
Au cours des études cliniques, des augmentations dose-dépendantes de la créatininémie ont été observées chez 36 % des patients, même si la plupart sont restées dans la limite supérieure de la normale. Des diminutions de la moyenne de la clairance de la créatinine ont été observées chez les patients pédiatriques et chez les patients adultes présentant une bêta-thalassémie avec une surcharge en fer durant la première année de traitement, mais il a été mis en évidence qu’il n’y a pas eu davantage de diminution au cours des années suivantes de traitement. Des élévations des transaminases hépatiques ont été rapportées. Des calendriers de surveillance de la sécurité des paramètres rénaux et hépatiques sont recommandés. Les troubles auditifs (diminution de l’audition) et oculaires (opacités du cristallin) sont peu fréquents, et des examens annuels sont également recommandés (voir rubrique 4.4).
Des effets indésirables cutanés graves (EICG) incluant des cas de syndrome de Stevens-Johnson (SSJ), de nécrolyse épidermique toxique (NET) et de syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) ont été rapportés après l’administration d’EXJADE (voir rubrique 4.4).
Tableau listant les effets indésirables
Les effets indésirables ci-dessous sont classés en utilisant la convention suivante : très fréquent (1/10) ; fréquent (1/100, <1/10) ; peu fréquent (1/1 000, <1/100) ; rare (1/10 000, <1/1 000) ; très rare (<1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 5
Affections hématologiques et du système lymphatique | ||
| Fréquence indéterminée : | Pancytopénie1, thrombopénie1, anémie aggravée1, neutropénie1 |
Affections du système immunitaire | ||
| Fréquence indéterminée : | Réactions d’hypersensibilité (y compris réactions anaphylactiques et angioedème)1 |
Troubles du métabolisme et de la nutrition | ||
| Fréquence indéterminée : | Acidose métabolique1 |
Affections psychiatriques | ||
| Peu fréquent : | Anxiété, troubles du sommeil |
Affections du système nerveux | ||
| Fréquent : | Céphalées |
| Peu fréquent : | Vertiges |
Affections oculaires | ||
| Peu fréquent : | Cataracte, maculopathie |
| Rare : | Névrite optique |
Affections de l’oreille et du labyrinthe | ||
| Peu fréquent : | Surdité |
Affections respiratoires, thoraciques et médiastinales | ||
| Peu fréquent : | Douleurs laryngées |
Affections gastro-intestinales | ||
| Fréquent : | Diarrhées, constipation, vomissements, nausées, douleurs abdominales, ballonnements, dyspepsie |
| Peu fréquent : | Hémorragie digestives, ulcère gastrique (y compris ulcères multiples), ulcères duodénaux, gastrite |
| Rare : | Oesophagite |
| Fréquence indéterminée : | Perforation gastro-intestinale1, pancréatite aiguë1 |
Affections hépatobiliaires | ||
| Fréquent : | Augmentation des transaminases |
| Peu fréquent : | Hépatite, lithiase biliaire |
| Fréquence indéterminée : | Insuffisance hépatique1, 2 |
Affections de la peau et du tissu sous-cutané | ||
| Fréquent : | Eruption cutanée, prurit |
| Peu fréquent : | Troubles de la pigmentation |
| Rare : | Syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) |
| Fréquence indéterminée : | Syndrome de Stevens-Johnson1, vascularites d’hypersensibilité1, urticaire1, érythème polymorphe1, alopécie1, nécrolyse épidermique toxique (NET)1 |
Affections du rein et des voies urinaires | ||
| Très fréquent : | Augmentation de la créatininémie |
| Fréquent : | Protéinurie |
| Peu fréquent : | Trouble de la fonction tubulaire rénale2 (syndrome de Fanconi acquis), glycosurie |
| Fréquence indéterminée : | Insuffisance rénale aiguë1, 2, néphrite tubulo-interstitielle1, lithiase rénale1, nécrose tubulaire rénale1 |
Troubles généraux et anomalies au site d’administration | ||
| Peu fréquent : | Pyrexie, œdème, fatigue |
1 Effets indésirables rapportés depuis la commercialisation d’EXJADE. Il s’agit de notifications spontanées pour lesquelles il est impossible de déterminer la fréquence ou la relation de causalité avec l’exposition au produit.
2 Des formes sévères associées à des modifications de l’état de conscience dans un contexte d’encéphalopathie hyperammoniémique ont été rapportées.
Description des effets indésirables sélectionnés
Des calculs biliaires et des troubles biliaires ont été rapportés chez environ 2 % des patients. Des augmentations des transaminases hépatiques ont été rapportées comme effet indésirable chez 2 % des patients. Des augmentations des transaminases supérieures à 10 fois la limite supérieure de la normale, suggérant une hépatite, ont été peu fréquentes (0,3 %). Depuis la commercialisation du déférasirox, des cas d’insuffisance hépatique, d’évolution parfois fatale, ont été rapportés (voir rubrique 4.4). Des cas d’acidose métabolique ont été rapportés depuis la commercialisation. La majorité de ces patients étaient atteints d’insuffisance rénale, de tubulopathie rénale (syndrome de Fanconi) ou de diarrhées, ou de pathologies ayant pour complication connue un déséquilibre acido-basique (voir rubrique 4.4). Des cas de pancréatite aiguë sévère ont été observés sans troubles biliaires sous-jacents confirmés. Comme avec les autres traitements chélateurs du fer, une perte d’audition des hautes fréquences et des opacités du cristallin (cataracte précoce) ont été peu fréquemment observées chez les patients traités par déférasirox (voir rubrique 4.4).
Clairance de la créatinine lors d’une surcharge en fer post-transfusionnelle
Dans une méta-analyse rétrospective menée sur 2102 patients bêta-thalassémiques adultes et enfants présentant une surcharge en fer post-transfusionnelle traités par déférasirox sous forme de comprimé dispersible dans deux études cliniques randomisées et quatre études en ouvert d’une durée pouvant atteindre 5 ans, une diminution moyenne de la clairance de la créatinine de 13,2 % chez les patients adultes (95 % IC : ‑14,4 % à ‑12,1 % ; n=935) et de 9,9 % chez les enfants (95 % IC : ‑11,1 % à ‑8,6 % ; n=1142) a été observée durant la première année de traitement. Chez 250 patients ayant été suivi jusqu’à 5 ans, aucune baisse ultérieure de la clairance moyenne de la créatinine n’a été observée.
Etude clinique chez les patients présentant un syndrome thalassémique non dépendant des transfusions
Au cours d’une étude d’un an chez des patients présentant des syndromes thalassémiques non dépendants des transfusions et une surcharge en fer (traités par les comprimés dispersibles à la posologie de 10 mg/kg/jour), les diarrhées (9,1 %), les éruptions cutanées (9,1 %) et les nausées (7,3 %) ont été les événements indésirables liés à la prise du médicament les plus fréquemment rapportés. Des anomalies des valeurs de la créatininémie et de la clairance de la créatinine ont été rapportées respectivement chez 5,5 % et 1,8 % des patients. Des élévations des transaminases hépatiques supérieures à 2 fois la valeur initiale et à 5 fois la limite supérieure de la normale ont été rapportées chez 1,8 % des patients.
Population pédiatrique
Dans deux études cliniques, la croissance et le développement sexuel des enfants traités par déférasirox pendant une durée allant jusqu’à 5 ans n’ont pas été altérés (voir rubrique 4.4).
La diarrhée est observée plus fréquemment chez les enfants âgés de 2 à 5 ans que chez les enfants plus âgés.
La tubulopathie rénale a été principalement observée chez les enfants et adolescents atteints de bêta-thalassémie traités par déférasirox. Depuis la mise sur le marché, une proportion élevée de cas d’acidose métabolique sont survenus chez des enfants atteints du syndrome de Fanconi.
Des pancréatites aiguës ont été rapportées, en particulier chez les enfants et les adolescents.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via:
Belgique
Agence fédérale des médicaments et des produits de santé
Division vigilance
Avenue Galilée 5/03
1210 Bruxelles
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet: www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Irlande
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EXJADE 90 mg comprimés pelliculés
EU/1/06/356/011
EU/1/06/356/012
EU/1/06/356/013
EXJADE 180 mg comprimés pelliculés
EU/1/06/356/014
EU/1/06/356/015
EU/1/06/356/016
EXJADE 360 mg comprimés pelliculés
EU/1/06/356/017
EU/1/06/356/018
EU/1/06/356/019
10. DATE DE MISE À JOUR DU TEXTE
28.09.2023
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne du médicament http://www.ema.europa.eu
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3540093 | EXJADE 90 MG COMP PELL 90 X 90 MG | V03AC03 | € 193,94 | - | Oui | - | - |
| 3540101 | EXJADE 180 MG COMP PELL 90 X 180 MG | V03AC03 | € 408,58 | - | Oui | - | - |
| 3540119 | EXJADE 360 MG COMP PELL 90 X 360 MG | V03AC03 | € 751,38 | - | Oui | - | - |