SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
Kadcyla 100 mg poeder voor concentraat voor oplossing voor infusie
Kadcyla 160 mg poeder voor concentraat voor oplossing voor infusie
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Kadcyla 100 mg poeder voor concentraat voor oplossing voor infusie
Eén injectieflacon poeder voor concentraat voor oplossing voor infusie bevat 100 mg trastuzumab-emtansine. Na reconstitutie bevat een injectieflacon van 5 ml oplossing 20 mg/ml trastuzumab-emtansine (zie rubriek 6.6).
Kadcyla 160 mg poeder voor concentraat voor oplossing voor infusie
Eén injectieflacon poeder voor concentraat voor oplossing voor infusie bevat 160 mg trastuzumab-emtansine. Na reconstitutie bevat een injectieflacon van 8 ml oplossing 20 mg/ml trastuzumab-emtansine (zie rubriek 6.6).
Hulpstoffen met bekend effect
Elke 100 mg injectieflacon bevat 1,38 mg natrium en 1,1 mg polysorbaat 20.
Elke 160 mg injectieflacon bevat 2,24 mg natrium en 1,7 mg polysorbaat 20.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
Trastuzumab-emtansine is een antilichaam-geneesmiddelconjugaat dat trastuzumab bevat, een gehumaniseerd monoklonaal IgG1-antilichaam dat wordt geproduceerd door een zoogdiercelsuspensiekweek (ovariumcellen van de Chinese hamster), covalent gebonden aan DM1, een microtubulineremmer, middels de stabiele thio-etherkoppeling MCC (4‑[N‑maleïmidomethyl]-cyclohexaan‑1‑carboxylaat).
3. FARMACEUTISCHE VORM
Poeder voor concentraat voor oplossing voor infusie.
Wit tot gebroken wit gelyofiliseerd poeder.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Vroege borstkanker
Kadcyla is als monotherapie geïndiceerd voor de adjuvante behandeling van volwassen patiënten met vroege HER2-positieve borstkanker die invasieve restziekte hebben in de borst en/of de lymfeklieren na een op taxaan gebaseerde en HER2-gerichte neoadjuvante behandeling.
Gemetastaseerde borstkanker
Kadcyla is als monotherapie geïndiceerd voor de behandeling van volwassen patiënten met HER2-positieve, niet-reseceerbare lokaal gevorderde of gemetastaseerde borstkanker die eerder trastuzumab en een taxaan, afzonderlijk of in combinatie, hebben ontvangen. Patiënten dienen:
eerdere therapie te hebben ontvangen voor lokaal gevorderde of gemetastaseerde ziekte, of
een recidief te hebben ontwikkeld tijdens of binnen zes maanden na het voltooien van adjuvante therapie.
4.2 Dosering en wijze van toediening
Kadcyla mag alleen worden voorgeschreven door een arts en worden toegediend middels intraveneuze infusie onder het toezicht van een beroepsbeoefenaar in de gezondheidszorg die ervaren is in de behandeling van patiënten met kanker (en dus in staat is om allergische/anafylactische infusiereacties te behandelen in een omgeving die zodanig is uitgerust dat reanimatie onmiddellijk kan plaatsvinden (zie rubriek 4.4)).
Patiënten die worden behandeld met trastuzumab-emtansine dienen een HER2-positieve tumorstatus te hebben, gedefinieerd als een immunohistochemie (IHC) -score van 3+ of een in-situ hybridisatie (ISH) of fluorescente in-situ hybridisatie (FISH) -ratio van ≥ 2,0, vastgesteld met behulp van een, van CE-markering voorzien, medisch apparaat voor in-vitrodiagnostiek (IVD). Indien geen CE-gemarkeerd IVD beschikbaar is dient de HER2-status te worden vastgesteld met behulp van een plaatsvervangende gevalideerde test.
Om medicatiefouten te voorkomen is het belangrijk om de injectieflaconetiketten te controleren, om er zeker van te zijn dat het geneesmiddel dat bereid en toegediend wordt Kadcyla (trastuzumab-emtansine) is en niet een ander trastuzumab-bevattend product (bijv. trastuzumab of trastuzumab‑deruxtecan).
Dosering
De aanbevolen dosering trastuzumab-emtansine is 3,6 mg/kg lichaamsgewicht, elke 3 weken (cyclus van 21 dagen) toegediend als een intraveneuze infusie.
De aanvangsdosis dient te worden toegediend als een 90 minuten durende intraveneuze infusie. Patiënten dienen tijdens de infusie en gedurende ten minste 90 minuten na de eerste infusie te worden gecontroleerd op koorts, rillingen of andere infusiegerelateerde reacties. De infusieplaats dient nauwlettend te worden gecontroleerd op mogelijke subcutane infiltratie tijdens de toediening. Gevallen van vertraagde epidermale schade of necrose na extravasatie zijn waargenomen na het op de markt brengen (zie rubriek 4.4 en 4.8).
Als de voorafgaande infusie goed werd verdragen kunnen daaropvolgende doses trastuzumab-emtansine worden toegediend als 30 minuten durende infusies. Patiënten dienen tijdens de infusie en gedurende ten minste 30 minuten na de infusie te worden geobserveerd.
De infusiesnelheid van trastuzumab-emtansine dient te worden vertraagd of de infusie dient te worden onderbroken indien de patiënt infusiegerelateerde symptomen ontwikkelt (zie rubriek 4.4 en 4.8). Bij levensbedreigende infusiereacties dient de toediening van trastuzumab-emtansine te worden gestaakt.
Duur van de behandeling
Vroege borstkanker
Patiënten moeten in totaal 14 cycli ontvangen, behalve als terugkeer van ziekte of onbehandelbare toxiciteit optreedt.
Gemetastaseerde borstkanker
Patiënten moeten behandeld worden tot ziekteprogressie of onbehandelbare toxiciteit optreedt.
Dosisaanpassing
Voor de behandeling van symptomatische bijwerkingen kan tijdelijke onderbreking, dosisverlaging of staken van de behandeling met trastuzumab-emtansine nodig zijn volgens de richtlijnen zoals vermeld in de tekst en Tabel 1 en 2.
De trastuzumab-emtansine-dosis mag niet meer worden verhoogd nadat een dosisverlaging is doorgevoerd.
Tabel 1 Schema voor dosisverlaging
Schema dosisverlaging | Toe te dienen dosis |
Eerste dosisverlaging | 3 mg/kg |
Tweede dosisverlaging | 2,4 mg/kg |
Noodzaak voor verdere dosisverlaging | Behandeling staken |
Tabel 2 Richtlijnen voor dosisaanpassing
Dosisaanpassingen bij patiënten met vroege borstkanker | ||
Bijwerking | Ernst | Aanpassing behandeling |
Trombocytopenie | Graad 2-3 op de dag van geplande behandeling | Dien geen trastuzumab-emtansine toe tot verbetering van trombocytenaantal tot graad ≤ 1 (≥ 75.000/mm3) en hervat vervolgens de behandeling met dezelfde dosis. |
Graad 4 op enig moment | Dien geen trastuzumab-emtansine toe tot verbetering van trombocytenaantal tot graad ≤ 1 (≥ 75.000/mm3) en verlaag vervolgens de dosis met één niveau. | |
Verhoogd alanineaminotransferase (ALAT) | Graad 2-3 | Dien geen trastuzumab-emtansine toe tot verbetering van ALAT tot graad ≤ 1 en verlaag vervolgens de dosis met één niveau. |
Graad 4 | Staak de behandeling met trastuzumab-emtansine. | |
Verhoogd aspartaataminotransferase (ASAT) | Graad 2 | Dien geen trastuzumab-emtansine toe tot verbetering van ASAT tot graad ≤ 1 en hervat vervolgens de behandeling met dezelfde dosis. |
Graad 3 | Dien geen trastuzumab-emtansine toe tot verbetering van ASAT tot graad ≤ 1 en verlaag vervolgens de dosis met één niveau. | |
Graad 4 | Staak de behandeling met trastuzumab-emtansine. | |
Hyperbilirubinemie | Totaalbilirubine | Dien geen trastuzumab-emtansine toe tot verbetering van bilirubine tot ≤ 1 × ULN en verlaag vervolgens de dosis met één niveau. |
Totaalbilirubine | Staak de behandeling met trastuzumab-emtansine. | |
Geneesmiddel-geïnduceerde leverschade (DILI) | Serumtransaminasen > 3 × ULN en gelijktijdig totaalbilirubine > 2 × ULN | Staak permanent de behandeling met trastuzumab-emtansine bij ontbreken van een andere aannemelijke oorzaak voor de verhoogde leverenzym- en bilirubinewaarden, zoals bijvoorbeeld levermetastasen of comedicatie. |
Nodulaire regeneratieve hyperplasie (NRH) | Elke graad | Staak permanent de behandeling met trastuzumab-emtansine. |
Perifere neuropathie | Graad 3-4 | Dien geen trastuzumab-emtansine toe tot verbetering tot graad ≤ 2. |
Linkerventrikeldisfunctie | LVEF < 45% | Dien geen trastuzumab-emtansine toe. Herhaal LVEF-bepaling binnen 3 weken. Als LVEF < 45% wordt bevestigd staak dan de behandeling met trastuzumab-emtansine. |
LVEF 45% tot < 50% met een afname van ≥ 10 procentpunten ten opzichte van baseline* | Dien geen trastuzumab-emtansine toe. Herhaal LVEF-bepaling binnen 3 weken. Als LVEF < 50% blijft en niet is hersteld tot < 10 procentpunten ten opzichte van baseline staak dan de behandeling met trastuzumab-emtansine. | |
LVEF 45% tot < 50% met een afname van < 10 procentpunten ten opzichte van baseline* | Continueer de behandeling met trastuzumab-emtansine. Herhaal LVEF-bepaling binnen 3 weken. | |
LVEF ≥ 50% | Continueer de behandeling met trastuzumab-emtansine. | |
Hartfalen | Symptomatisch CHF, graad 3-4 LVSD of graad 3-4 hartfalen, of graad 2 hartfalen gepaard gaand met LVEF < 45% | Staak de behandeling met trastuzumab-emtansine. |
Pulmonale toxiciteit | Interstitiële longziekte (ILD) of pneumonitis | Staak permanent de behandeling met trastuzumab-emtansine. |
Bestralingsgerelateerde pneumonitis | Graad 2 | Staak de behandeling met trastuzumab-emtansine als standaardbehandeling geen verbetering biedt. |
Graad 3-4 | Staak de behandeling met trastuzumab-emtansine. | |
Dosisaanpassingen bij patiënten met gemetastaseerde borstkanker | ||
Bijwerking | Ernst | Aanpassing behandeling |
Trombocytopenie | Graad 3 | Dien geen trastuzumab-emtansine toe tot verbetering van trombocytenaantal tot graad ≤ 1 (≥ 75.000/mm3) en hervat vervolgens de behandeling met dezelfde dosis. |
Graad 4 | Dien geen trastuzumab-emtansine toe tot verbetering van trombocytenaantal tot graad ≤ 1 (≥ 75.000/mm3) en verlaag vervolgens de dosis met één niveau. | |
Verhoogd transaminase (ASAT/ALAT) | Graad 2 | Behandel met dezelfde dosis. |
Graad 3 | Dien geen trastuzumab-emtansine toe tot verbetering van ASAT/ALAT tot graad ≤ 2 en verlaag vervolgens de dosis met één niveau. | |
Graad 4 | Staak de behandeling met trastuzumab-emtansine. | |
Hyperbilirubinemie | Graad 2 | Dien geen trastuzumab-emtansine toe tot verbetering van totaal bilirubine tot graad ≤ 1 en hervat vervolgens de behandeling met dezelfde dosis. |
Graad 3 | Dien geen trastuzumab-emtansine toe tot verbetering van totaal bilirubine tot graad ≤ 1 en verlaag vervolgens de dosis met één niveau. | |
Graad 4 | Staak de behandeling met trastuzumab-emtansine. | |
Geneesmiddel-geïnduceerde leverschade (DILI) | Serumtransaminasen > 3 × ULN en gelijktijdig totaal bilirubine > 2 × ULN | Staak permanent de behandeling met trastuzumab-emtansine bij ontbreken van een andere aannemelijke oorzaak voor de verhoogde leverenzym- en bilirubinewaarden, zoals levermetastasen of comedicatie. |
Nodulaire regeneratieve hyperplasie (NRH) | Elke graad | Staak permanent de behandeling met trastuzumab-emtansine. |
Linkerventrikeldisfunctie | Symptomatisch CHF | Staak de behandeling met trastuzumab-emtansine. |
LVEF < 40% | Dien geen trastuzumab-emtansine toe. Herhaal LVEF-bepaling binnen 3 weken. Als LVEF < 40% bevestigd wordt staak dan de behandeling met trastuzumab-emtansine. | |
LVEF 40% tot ≤ 45% met een afname van ≥ 10 procentpunten ten opzichte van baseline* | Dien geen trastuzumab-emtansine toe. Herhaal LVEF-bepaling binnen 3 weken. Als LVEF niet is hersteld tot < 10 procentpunten ten opzichte van baseline staak dan de behandeling met trastuzumab-emtansine. | |
LVEF 40% tot ≤ 45% met een afname van < 10 procentpunten ten opzichte van baseline* | Continueer de behandeling met trastuzumab-emtansine. Herhaal LVEF-bepaling binnen 3 weken. | |
LVEF > 45% | Continueer de behandeling met trastuzumab-emtansine. | |
Perifere neuropathie | Graad 3-4 | Dien geen trastuzumab-emtansine toe tot verbetering tot graad ≤ 2. |
Pulmonale toxiciteit | Interstitiële longziekte (ILD) of pneumonitis | Staak permanent de behandeling met trastuzumab-emtansine. |
ALAT = alanineaminotransferase; ASAT = aspartaataminotransferase; CHF = congestief hartfalen; LVEF = linkerventrikelejectiefractie; LVSD = linkerventrikel systolische linkerventrikeldisfunctie; ULN = bovengrens van normaal
* Voorafgaand aan de behandeling met trastuzumab-emtansine.
Uitgestelde of gemiste dosis
Indien een geplande dosis is gemist, dan dient deze zo spoedig mogelijk alsnog te worden toegediend; zonder te wachten op de volgende geplande cyclus. Het toedieningsschema dient te worden aangepast om een 3-wekelijks interval tussen de doses te handhaven. De volgende dosis dient in overeenstemming met de bovenstaande doseringsaanbevelingen te worden toegediend.
Perifere neuropathie
De behandeling met trastuzumab-emtansine dient tijdelijk te worden gestaakt bij patiënten die perifere neuropathie graad 3 of 4 ondervinden, totdat deze is verbeterd tot ≤ graad 2. Bij het herstarten van de behandeling kan een dosisverlaging worden overwogen volgens het schema voor dosisverlaging (zie Tabel 1).
Speciale populaties
Ouderen
Er is geen dosisaanpassing nodig bij patiënten van ≥ 65 jaar oud. Er zijn onvoldoende gegevens om de veiligheid en werkzaamheid bij patiënten van ≥ 75 jaar oud vast te stellen vanwege beperkte gegevens in deze subgroep. Een subgroepanalyse van 345 patiënten van ≥ 65 jaar uit onderzoek MO28231 toonde echter een tendens naar een hogere incidentie van bijwerkingen van graad 3, 4 en 5, ernstige bijwerkingen en bijwerkingen die leidden tot het tijdelijk of permanent stopzetten van de behandeling, maar een vergelijkbare incidentie van bijwerkingen van graad 3 of hoger die beschouwd werden als gerelateerd aan de behandeling.
De populatiefarmacokinetische analyse geeft aan dat leeftijd geen klinisch relevant effect heeft op de farmacokinetiek van trastuzumab-emtansine (zie rubriek 5.1 en 5.2).
Verminderde nierfunctie
Bij patiënten met een licht of matig verminderde nierfunctie is aanpassing van de startdosis niet nodig (zie rubriek 5.2). De mogelijke noodzaak tot dosisaanpassing bij patiënten met een ernstig verminderde nierfunctie kan niet worden vastgesteld omdat er onvoldoende gegevens beschikbaar zijn. Patiënten met een ernstig verminderde nierfunctie dienen daarom zorgvuldig te worden gecontroleerd.
Verminderde leverfunctie
Er is geen aanpassing van de startdosis nodig voor patiënten met een licht of matig verminderde leverfunctie. Trastuzumab-emtansine is niet onderzocht in patiënten met een ernstig verminderde leverfunctie. Voorzichtigheid moet worden betracht bij behandeling van patiënten met een verminderde leverfunctie vanwege de bekende hepatotoxiciteit die wordt gezien met trastuzumab-emtansine (zie rubriek 4.4 en 5.2).
Pediatrische patiënten
De veiligheid en werkzaamheid bij kinderen en jongeren tot 18 jaar zijn niet vastgesteld omdat er geen relevante toepassing is bij pediatrische patiënten voor de indicatie borstkanker.
Wijze van toediening
Kadcyla is bedoeld voor intraveneus gebruik. Trastuzumab-emtansine moet worden gereconstitueerd en verdund door een beroepsbeoefenaar in de gezondheidszorg en worden toegediend als een intraveneuze infusie. Het mag niet worden toegediend als een intraveneuze push- of bolusinjectie.
Voor instructies over reconstitutie en verdunning van het geneesmiddel voorafgaand aan toediening, zie rubriek 6.6.
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
4.8 Bijwerkingen
Overzicht van het veiligheidsprofiel
De veiligheid van trastuzumab-emtansine is in klinische onderzoeken onderzocht bij 2.611 borstkankerpatiënten. In deze patiëntenpopulatie waren:
• de meest voorkomende ernstige bijwerkingen (> 0,5% van de patiënten) bloedingen, pyrexie, trombocytopenie, dyspneu, buikpijn, musculoskeletale pijn en braken.
• de meest voorkomende bijwerkingen (≥ 25%) van trastuzumab-emtansine misselijkheid, vermoeidheid, musculoskeletale pijn, bloedingen, hoofdpijn, verhoogde transaminasespiegels, trombocytopenie en perifere neuropathie. De meerderheid van de gemelde bijwerkingen waren graad 1 of 2 in ernst.
• de meest voorkomende graad ≥ 3 bijwerkingen volgens de Common Terminology Criteria for Adverse Events van het National Cancer Institute (NCI-CTCAE) (> 2%) trombocytopenie, verhoogde transaminasespiegels, anemie, neutropenie, vermoeidheid en hypokaliëmie.
Lijst van bijwerkingen in tabelvorm
De bijwerkingen van 2.611 patiënten die werden behandeld met trastuzumab-emtansine staan vermeld in Tabel 3. De bijwerkingen staan hieronder weergegeven op basis van de systeem/orgaanklasse (SOC) en frequentiecategorieën volgens MedDRA. De frequentiecategorieën zijn gedefinieerd als zeer vaak (≥ 1/10), vaak (≥ 1/100, < 1/10), soms (≥ 1/1.000, < 1/100), zelden (≥ 1/10.000, < 1/1.000), zeer zelden (< 1/10.000) en niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen elke frequentiecategorie en SOC worden de bijwerkingen weergegeven in volgorde van afnemende ernst. Bijwerkingen werden gemeld met behulp van NCI-CTCAE voor de beoordeling van de toxiciteit.
Tabel 3 Lijst van bijwerkingen bij patiënten die werden behandeld met trastuzumab-emtansine in klinische onderzoeken
Systeem/orgaanklasse | Frequentie | Bijwerking |
Infecties en parasitaire aandoeningen | Zeer vaak | Urineweginfectie |
Bloed- en lymfestelselaandoeningen | Zeer vaak | Trombocytopenie, Anemie, |
Vaak | Neutropenie, Leukopenie | |
Immuunsysteemaandoeningen | Vaak | Geneesmiddel-overgevoeligheid |
Voedings- en stofwisselingsstoornissen | Vaak | Hypokaliëmie |
Psychische stoornissen | Zeer vaak | Slapeloosheid |
Zenuwstelselaandoeningen | Zeer vaak | Perifere neuropathie, Hoofdpijn |
Vaak | Duizeligheid, Dysgeusie, Geheugenstoornis | |
Oogaandoeningen | Vaak | Droge ogen, Conjunctivitis, Wazig zien, Toegenomen traanproductie |
Hartaandoeningen | Vaak | Linkerventrikeldisfunctie |
Bloedvataandoeningen | Zeer vaak | Bloedingen |
Vaak | Hypertensie | |
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen | Zeer vaak | Epistaxis, Hoesten, Dyspneu |
Soms | Pneumonitis (ILD) | |
Maagdarmstelselaandoeningen | Zeek vaak | Stomatitis, Diarree, Braken, Misselijkheid, Constipatie, Droge mond, Buikpijn |
Vaak | Dyspepsie, Tandvleesbloeding | |
Lever- en galaandoeningen | Zeer vaak | Verhoogde transaminasespiegels |
Vaak | Alkalische fosfatase in bloed verhoogd, Bilirubine in bloed verhoogd | |
Soms | Levertoxiciteit, Leverfalen, Nodulaire regeneratieve hyperplasie, Portale hypertensie | |
Zelden | Nierfalen | |
Huid- en onderhuidaandoeningen | Vaak | Huiduitslag, Pruritus, Alopecia, Nagelaandoening, Palmoplantair erytrodysesthesie-syndroom, Urticaria |
Skeletspierstelsel- en bindweefselaandoeningen | Zeer vaak | Musculoskeletale pijn, Artralgie, Myalgie |
Algemene aandoeningen en toedieningsplaatsstoornissen | Zeer vaak | Vermoeidheid, Pyrexie, Asthenie |
Vaak | Perifeer oedeem, Rillingen | |
Soms | Extravasatie op de injectieplaats | |
Letsels, intoxicaties en verrichtingscomplicaties | Vaak | Infusiegerelateerde reacties |
Soms | Bestralingsgerelateerde pneumonitis |
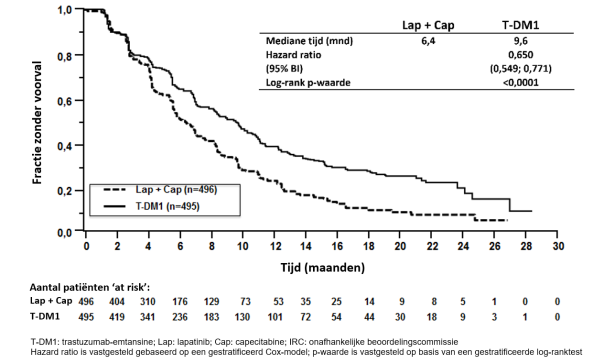
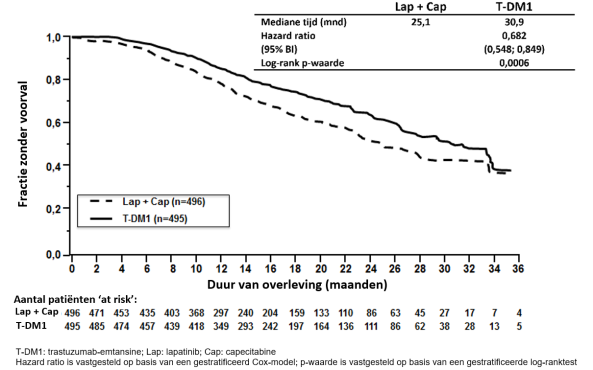
Tabel 3 geeft de samengevoegde data weer van de totale behandelingsperiode in de onderzoeken naar gemetastaseerde borstkanker (N = 1. 871; mediane aantal cycli van trastuzumab-emtansine was 10) en KATHERINE (N = 740; mediane aantal cycli van trastuzumab-emtansine was 14).
Beschrijving van geselecteerde bijwerkingen
Trombocytopenie
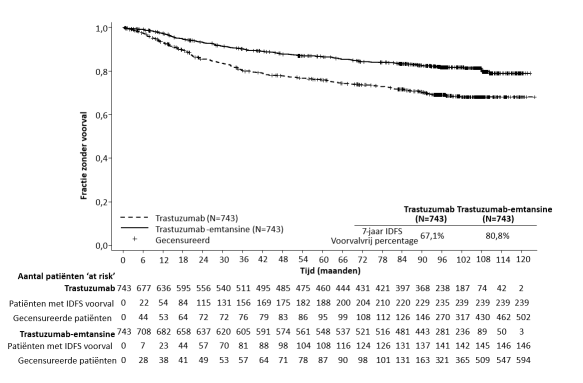
Trombocytopenie, of verlaagd aantal trombocyten, werd gemeld bij 24,9% van de patiënten in klinische onderzoeken in gemetastaseerde borstkanker met trastuzumab-emtansine en was de meest voorkomende bijwerking die leidde tot het staken van de behandeling (2,6%). Trombocytopenie werd gemeld bij 28,6% van de patiënten met vroege borstkanker in klinische onderzoeken met trastuzumab-emtansine en was de meest gemelde bijwerking voor alle graden en graad ≥ 3, alsook de meest gemelde bijwerking die leidde tot het staken van de behandeling (4,2%), dosisonderbreking en dosisverlaging. De meeste patiënten hadden voorvallen van graad 1 of 2 (≥ 50.000/mm3), met het dieptepunt op dag 8. In het algemeen verbeterde dit naar graad 0 of 1 (≥ 75.000/mm3) ten tijde van de volgende geplande dosis. In klinische onderzoeken waren de incidentie en ernst van trombocytopenie hoger bij Aziatische patiënten. Onafhankelijk van het ras was de incidentie van voorvallen van graad 3 of 4 (< 50.000/mm3) 8,7% bij patiënten met gemetastaseerde borstkanker die werden behandeld met trastuzumab-emtansine en 5,7% bij patiënten met vroege borstkanker. Voor dosisaanpassingen bij trombocytopenie, zie rubriek 4.2 en 4.4.
Bloedingen
Hemorragische voorvallen werden gemeld bij 34,8% van de patiënten met gemetastaseerde borstkanker in klinische onderzoeken met trastuzumab-emtansine en de incidentie van ernstige hemorragische voorvallen (graad ≥ 3) was 2,2%. Hemorragische voorvallen werden gemeld in bij 29,2% van de patiënten met vroege borstkanker en de incidentie van ernstige hemorragische voorvallen (graad ≥ 3) was 0,4%, waaronder één voorval van graad 5. In sommige van de waargenomen gevallen hadden de patiënten trombocytopenie of kregen de patiënten ook anticoagulantia of antitrombocytenbehandeling; in andere gevallen waren er geen andere bekende risicofactoren. Gevallen van bloedingen met fatale afloop zijn waargenomen bij zowel patiënten met gemetastaseerde borstkanker als vroege borstkanker.
Verhoogde transaminasespiegels (ASAT/ALAT)
In klinische onderzoeken is een toename van de concentraties serumtransaminasen (graad 1-4) waargenomen tijdens behandeling met trastuzumab-emtansine (zie rubriek 4.4). De verhogingen in transaminasespiegels waren in het algemeen van voorbijgaande aard. Een cumulatief effect van trastuzumab-emtansine op transaminasespiegels is waargenomen en over het algemeen trad herstel op als de behandeling werd gestaakt. Verhoogde transaminasespiegels werden gemeld bij 24,2% van de patiënten met gemetastaseerde borstkanker in klinische onderzoeken. Verhoogde ASAT en ALAT van graad 3 of 4 werden gemeld bij respectievelijk 4,2% en 2,7% van de patiënten met gemetastaseerde borstkanker en deed zich meestal voor tijdens de vroege behandelingscycli (1-6). Verhoogde transaminasespiegels werden gemeld bij 32,6% van de patiënten met vroege borstkanker. Verhoogde transaminasespiegels van graad 3 of 4 werden gemeld bij 1,6% van de patiënten met vroege borstkanker. Over het algemeen waren de hepatische voorvallen van graad ≥ 3 niet geassocieerd met een slechte klinische uitkomst; daaropvolgende follow-up waarden hadden de neiging verbetering te vertonen naar een bereik waardoor de patiënt in het onderzoek kon blijven en studiemedicatie kon blijven ontvangen met dezelfde of een verlaagde dosis. Er werd geen relatie waargenomen tussen de blootstelling aan trastuzumab-emtansine (AUC), de maximale serumconcentratie van trastuzumab-emtansine (Cmax), de totale blootstelling aan trastuzumab (AUC) of de Cmax van DM1 en verhogingen in transaminasespiegels. Voor dosisaanpassingen in het geval van verhoogde transaminasespiegels, zie rubriek 4.2 en 4.4.
Linkerventrikeldisfunctie
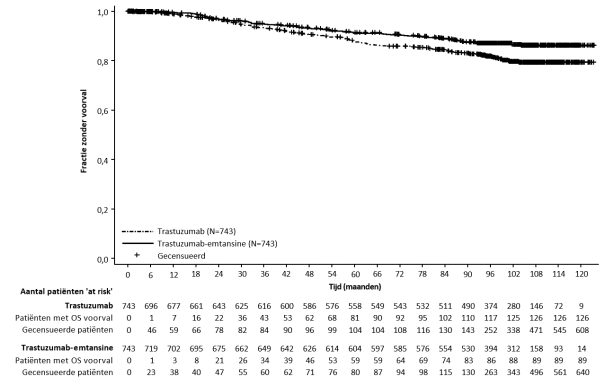
Linkerventrikeldisfunctie werd gemeld bij 2,2% van de patiënten met gemetastaseerde borstkanker in klinische onderzoeken met trastuzumab-emtansine. De meeste voorvallen betroffen een asymptomatische LVEF-daling van graad 1 of 2. Voorvallen van graad 3 of 4 werden gemeld bij 0,4% van de patiënten met gemetastaseerde borstkanker. In een observationeel onderzoek (BO39807), waarbij patiënten met gemetastaseerde borstkanker met een LVEF-aanvangswaarde van 40-49% startten met trastuzumab-emtansine, had ongeveer 22% (7 van de 32) van de patiënten met gemetastaseerde borstkanker een LVEF-afname van > 10% ten opzichte van de beginwaarde en/of CHF; de meerderheid van deze patiënten had andere cardiovasculaire risicofactoren. Linkerventrikeldisfunctie kwam voor bij 3,0% van de patiënten met vroege borstkanker, waarbij graad 3 bij 0,5% van de patiënten voorkwam en geen voorvallen van een hogere graad. Voor dosisaanpassingen in geval van LVEF-daling zie Tabel 2 in rubriek 4.2 en rubriek 4.4.
Perifere neuropathie
Perifere neuropathie, voornamelijk graad 1 en overwegend sensorisch, werd gemeld in klinische onderzoeken met trastuzumab-emtansine. Bij patiënten met gemetastaseerde borstkanker was de totale incidentie van perifere neuropathie 29,0% en 8,6% voor graad ≥ 2. Bij patiënten met vroege borstkanker was de totale incidentie van perifere neuropathie 32,0% en 10,1% voor graad ≥ 2.
Infusiegerelateerde reacties
Infusiegerelateerde reacties worden gekenmerkt door één of meer van de volgende symptomen: blozen, rillingen, pyrexie, dyspneu, hypotensie, piepende ademhaling, bronchospasme en tachycardie. Infusiegerelateerde reacties werden gemeld bij 4,0% van de patiënten met gemetastaseerde borstkanker in klinische onderzoeken met trastuzumab-emtansine, waarbij zes voorvallen van graad 3 en geen voorvallen van graad 4 werden gemeld. Infusiegerelateerde reacties werden gemeld bij 1,6% van de patiënten met vroege borstkanker, waarbij geen voorvallen van graad 3 of 4 werden gemeld. Infusiegerelateerde reacties verdwenen in de loop van enkele uren tot een dag na het beëindigen van de infusie. Er werd in klinische onderzoeken geen dosisrelatie waargenomen. Voor dosisaanpassingen in het geval van infusiegerelateerde reacties, zie rubriek 4.2 en 4.4.
Overgevoeligheidsreacties
Overgevoeligheid werd gemeld bij 2,6% van de patiënten met gemetastaseerde borstkanker in klinische onderzoeken met trastuzumab-emtansine, waarbij één voorval van graad 3 en één van graad 4 werden gemeld. Overgevoeligheid werd gemeld bij 2,7% van de patiënten met vroege borstkanker, waarbij voorvallen van graad 3 bij 0,4% van de patiënten werd gemeld en geen voorvallen met een hogere graad. Over het algemeen waren de meeste overgevoeligheidsreacties licht of matig van ernst en verdwenen na behandeling. Voor dosisaanpassingen in het geval van overgevoeligheidsreacties, zie rubriek 4.2 en 4.4.
Immunogeniciteit
Zoals met alle therapeutische eiwitten bestaat er een kans op een immuunrespons tegen trastuzumab-emtansine. Een totaal van 1.243 patiënten uit zeven klinische onderzoeken werd op meerdere tijdpunten getest op door de behandeling ontstane antilichamen (anti-drug antibodies, ADA) tegen trastuzumab-emtansine. Na toediening van trastuzumab-emtansine testte 5,1% (64/1.243) van de patiënten positief voor anti-trastuzumab-emtansineantilichamen op één of meer tijdpunten na de toediening. In de fase I- en fase II-onderzoeken testte 6,4% (24/376) van de patiënten positief voor anti-trastuzumab-emtansineantilichamen. In het EMILIA-onderzoek (TDM4370g/BO21977) testte 5,2% (24/466) van de patiënten positief voor anti-trastuzumab-emtansineantilichamen, waarbij 13 ook positief waren voor neutraliserende antilichamen. In het KATHERINE-onderzoek (BO27938) testte 4,0% (16/401) van de patiënten positief voor anti-trastuzumab-emtansineantilichamen, waarbij 5 ook positief waren voor neutraliserende antilichamen. Omdat antilichamen tegen het geneesmiddel weinig voorkomen, is het effect van deze antilichamen op de farmacokinetiek, farmacodynamiek, veiligheid en/of werkzaamheid van trastuzumab-emtansine onbekend.
Extravasatie
Reacties die secundair zijn aan extravasatie zijn waargenomen in klinische onderzoeken met trastuzumab-emtansine. Deze reacties waren meestal licht of matig ernstig en omvatten erytheem, gevoeligheid, huidirritatie, pijn of zwelling op de infusieplaats. Deze reacties zijn vaker waargenomen binnen 24 uur na de infusie. Na het op de markt brengen zijn incidentele gevallen van epidermale schade of necrose na extravasatie waargenomen binnen enkele dagen tot weken na de infusie. Een specifieke behandeling van trastuzumab-emtansine-extravasatie is op dit moment onbekend (rubriek 4.4).
Laboratoriumafwijkingen
Tabel 4 en 5 geven de laboratoriumafwijkingen weer die werden waargenomen bij patiënten die met trastuzumab-emtansine werden behandeld in de klinische onderzoeken TDM4370g/BO21977/EMILIA en BO27938/KATHERINE.
Tabel 4 Laboratoriumafwijkingen waargenomen bij patiënten die met trastuzumab-emtansine werden behandeld in onderzoek TDM4370g/BO21977/EMILIA
Parameter | Trastuzumab-emtansine (n = 490) | ||
Alle graden (%) | Graad 3 (%) | Graad 4 (%) | |
Lever | |||
Verhoogd bilirubine | 21 | < 1 | 0 |
Verhoogd ASAT | 98 | 8 | < 1 |
Verhoogd ALAT | 82 | 5 | < 1 |
Hematologie | |||
Verlaagd aantal trombocyten | 85 | 14 | 3 |
Verlaagd hemoglobine | 63 | 5 | 1 |
Verlaagd aantal neutrofielen | 41 | 4 | < 1 |
Kalium | |||
Verlaagd kalium | 35 | 3 | < 1 |
Tabel 5 Laboratoriumafwijkingen waargenomen bij patiënten die met trastuzumab-emtansine werden behandeld in onderzoek BO27938/KATHERINE
Parameter | Trastuzumab-emtansine (n=740) | ||
Alle graden (%) | Graad 3 (%) | Graad 4 (%) | |
Lever | |||
Verhoogd bilirubine | 11 | 0 | 0 |
Verhoogd ASAT | 79 | ≤ 1 | 0 |
Verhoogd ALAT | 55 | < 1 | 0 |
Hematologie | |||
Verlaagd aantal trombocyten | 51 | 4 | 2 |
Verlaagd hemoglobine | 31 | 1 | 0 |
Verlaagd aantal neutrofielen | 24 | 1 | 0 |
Kalium | |||
Verlaagd kalium | 26 | 2 | < 1 |
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden (zie hieronder voor details).
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Duitsland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/13/885/001
EU/1/13/885/002
10. DATUM VAN HERZIENING VAN DE TEKST
20 februari 2025
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3084670 | KADCYLA 160 MG PDR CONCENTR OPL INF 20 MG/ML | L01FD03 | - | € 2217,26 | Ja | - | - |
| 3084688 | KADCYLA 100 MG PDR CONCENTR OPL INF 20 MG/ML | L01FD03 | - | € 1385,79 | Ja | - | - |