BIJLAGE I
SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
Praluent 75 mg oplossing voor injectie in een voorgevulde pen
Praluent 150 mg oplossing voor injectie in een voorgevulde pen
Praluent 75 mg oplossing voor injectie in een voorgevulde spuit
Praluent 150 mg oplossing voor injectie in een voorgevulde spuit
Praluent 300 mg oplossing voor injectie in een voorgevulde pen
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Praluent 75 mg oplossing voor injectie in een voorgevulde pen
Elke voorgevulde pen voor eenmalig gebruik bevat 75 mg alirocumab in 1 ml oplossing.
Praluent 75 mg oplossing voor injectie in een voorgevulde spuit
Elke voorgevulde spuit voor eenmalig gebruik bevat 75 mg alirocumab in 1 ml oplossing.
Praluent 150 mg oplossing voor injectie in een voorgevulde pen
Elke voorgevulde pen voor eenmalig gebruik bevat 150 mg alirocumab in 1 ml oplossing.
Praluent 150 mg oplossing voor injectie in een voorgevulde spuit
Elke voorgevulde spuit voor eenmalig gebruik bevat 150 mg alirocumab in 1 ml oplossing.
Praluent 300 mg oplossing voor injectie in een voorgevulde pen
Elke voorgevulde pen voor eenmalig gebruik bevat 300 mg alirocumab in 2 ml oplossing.
Alirocumab is een humaan IgG1 monoklonaal antilichaam dat via recombinant-DNA-technologie in ovariumcellen van Chinese hamsters wordt geproduceerd.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Oplossing voor injectie (injectie).
Heldere, kleurloze tot lichtgele oplossing.
pH: 5,7 – 6,3
Osmolaliteit:
Praluent 75 mg oplossing voor injectie
293 – 439 mOsm/kg
Praluent 150 mg oplossing voor injectie
383 – 434 mOsm/kg
Praluent 300 mg oplossing voor injectie
383 – 434 mOsm/kg
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Primaire hypercholesterolemie en gemengde dyslipidemie
Praluent is geïndiceerd bij volwassenen met primaire hypercholesterolemie (heterozygoot familiair en niet-familiair) of gemengde dyslipidemie, en bij pediatrische patiënten van 8 jaar en ouder met heterozygote familiaire hypercholesterolemie (HeFH) ter aanvulling op een dieet:
in combinatie met een statine of een statine met andere lipidenverlagende therapieën bij patiënten bij
wie de LDL-C-streefwaarden niet bereikt worden met maximaal verdraagbare doseringen van een statine of
- alleen of in combinatie met andere lipidenverlagende therapieën bij patiënten die statine-intolerant
zijn, of voor wie een statine gecontra-indiceerd is.
Vastgestelde atherosclerotische cardiovasculaire ziekte
Praluent is geïndiceerd voor gebruik bij volwassenen met vastgestelde atherosclerotische cardiovasculaire ziekte om het cardiovasculair risico te verminderen door de LDL-C-waarden te verlagen, als aanvulling op de correctie van andere risicofactoren:
- in combinatie met de maximaal verdragen dosis van een statine, met of zonder andere lipidenverlagende behandelingen, of
- apart of in combinatie met andere lipidenverlagende behandelingen bij patiënten die statine-intolerant zijn of bij wie een statine gecontra-indiceerd is.
Raadpleeg rubriek 5.1 voor de onderzoeksresultaten met betrekking tot de effecten op LDL-C, cardiovasculaire voorvallen en onderzochte populaties.
4.2 Dosering en wijze van toediening
Dosering
Volwassenen
Voordat met alirocumab wordt begonnen, dienen secundaire oorzaken van hyperlipidemie of gemengde dyslipidemie (bijv. nefrotisch syndroom, hypothyreoïdie) te worden uitgesloten.
De gebruikelijke startdosering voor alirocumab is 75 mg, om de 2 weken subcutaan toegediend. Patiënten die een sterkere LDL-C-verlaging (>60%) nodig hebben, kunnen beginnen met 150 mg, om de 2 weken, of 300 mg om de 4 weken (maandelijks), subcutaan toegediend.
De dosering van alirocumab kan worden aangepast op basis van individuele patiëntkenmerken, zoals baseline LDL-C-waarde, behandeldoel en respons. De lipidenconcentraties kunnen 4 tot 8 weken na aanvang van de behandeling of aanpassing van de dosering worden beoordeeld. Daarna kan de dosering dienovereenkomstig worden aangepast (titratie omhoog of omlaag). Wanneer bijkomende LDL-C verlaging nodig is bij patiënten met 75 mg om de 2 weken of 300 mg om de 4 weken (maandelijks), kan de dosering aangepast worden tot de maximale dosering van 150 mg om de 2 weken.
HeFH bij pediatrische patiënten van 8 jaar en ouder
Lichaamsgewicht van patiënten | Aanbevolen dosis | Aanbevolen dosis als bijkomende LDL-C-verlaging nodig is* |
Minder dan 50 kg | 150 mg om de 4 weken | 75 mg om de 2 weken |
50 kg of meer | 300 mg om de 4 weken | 150 mg om de 2 weken |
* De lipidenniveaus kunnen 8 weken na opstarten of titratie van de behandeling beoordeeld worden en de dosis kan dienovereenkomstig worden aangepast.
Gemiste dosis
Indien een dosis Praluent wordt gemist, dient de dosis zo spoedig mogelijk te worden toegediend en daarna dient de dosering te worden hervat volgens het originele schema.
Speciale populaties
Ouderen
Er is geen aangepaste dosering nodig bij ouderen.
Leverinsufficiëntie
Er is geen aangepaste dosering nodig voor patiënten met lichte of matig ernstige leverinsufficiëntie. Er zijn geen gegevens beschikbaar over patiënten met ernstige leverinsufficiëntie (zie rubriek 5.2).
Nierinsufficiëntie
Er is geen aangepaste dosering nodig voor patiënten met lichte of matig ernstige nierinsufficiëntie. Er zijn beperkte gegevens beschikbaar over patiënten met ernstige nierinsufficiëntie (zie rubriek 5.2).
Lichaamsgewicht
Er is geen aangepaste dosering nodig bij patiënten op basis van gewicht.
Pediatrische patiënten
De veiligheid en werkzaamheid van Praluent bij kinderen onder de 8 jaar zijn nog niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Subcutaan gebruik.
Alirocumab wordt geïnjecteerd als subcutane injectie in de dij, buik of bovenarm.
Elke voorgevulde pen of voorgevulde spuit is uitsluitend voor eenmalig gebruik
De toediening van de 300 mg dosis gebeurt ofwel in 1 injectie van 300 mg ofwel in 2 opeenvolgende injecties van 150 mg op twee verschillende injectieplaatsen.
Het wordt aanbevolen om bij elke injectie van injectieplaats te wisselen.
Alirocumab mag niet worden geïnjecteerd in gebieden met een actieve huidaandoening of letsel zoals zonnebrand, huiduitslag, ontsteking of huidinfecties.
Alirocumab mag niet gelijktijdig met andere injecteerbare geneesmiddelen worden toegediend op dezelfde injectieplaats.
Voorzorgsmaatregelen vóór gebruik of toediening van het geneesmiddel
Laat de oplossing voorafgaand aan gebruik opwarmen tot kamertemperatuur (zie rubriek 6.6).
Pediatrische patiënten van 8 jaar en ouder
Bij jongeren van 12 jaar en ouder wordt het aanbevolen om Praluent toe te dienen door of onder het toezicht van een volwassene.
Bij kinderen jonger dan 12 jaar moet Praluent worden toegediend door een verzorger.
Volwassenen
Alirocumab kan door volwassen patiënten zelf of door een verzorger worden geïnjecteerd, nadat door een zorgverlener instructies over de juiste subcutane injectietechniek zijn gegeven.
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
De vaakst voorkomende bijwerkingen bij de aanbevolen dosering zijn: lokale reacties op de injectieplaats (6,1%), verschijnselen en symptomen van de bovenste luchtwegen (2,0%) en pruritus (1,1%). De vaakst voorkomende bijwerkingen die leidden tot stopzetting van de behandeling bij met alirocumab behandelde patiënten waren lokale reacties op de injectieplaats.
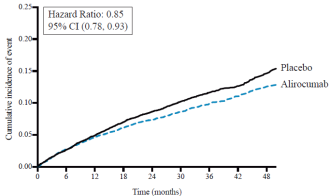
Het veiligheidsprofiel in ODYSSEY OUTCOMES kwam overeen met het totale veiligheidsprofiel dat in de gecontroleerde fase 3-onderzoeken werd beschreven.
Er werd geen verschil in het veiligheidsprofiel waargenomen tussen de twee doseringen (75 mg en 150 mg) gebruikt in het fase 3-programma.
Lijst van bijwerkingen in tabelvorm
De volgende bijwerkingen werden gemeld in samengevoegde gecontroleerde onderzoeken bij met alirocumab behandelde patiënten en/of postmarketinggebruik (zie tabel 1).
De frequentie van alle bijwerkingen waargenomen in klinische onderzoeken is berekend op basis van de incidentie in samengevoegde klinische fase 3-onderzoeken. De bijwerkingen zijn gerangschikt naar systeem/orgaanklasse. De frequentiecategorieën zijn als volgt gedefinieerd: zeer vaak (≥1/10); vaak (≥1/100 tot <1/10); soms (≥1/1.000 tot <1/100); zelden (≥1/10.000 tot <1/1.000); zeer zelden (<1/10.000) en niet bekend (kan met de beschikbare gegevens niet worden bepaald).
De frequentie van bijwerkingen gemeld tijdens postmarketinggebruik kan niet worden bepaald, omdat ze afkomstig zijn van spontane rapporten. Bijgevolg wordt de frequentie van deze bijwerkingen gekwalificeerd als “niet bekend”.
Tabel 1 – Bijwerkingen
Systeem/orgaanklasse | Vaak | Zelden | Niet bekend |
Immuunsysteemaandoeningen |
| Overgevoeligheid, |
|
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen | Verschijnselen en symptomen van de bovenste luchtwegen* |
|
|
Huid- en onderhuidaandoeningen | Pruritus | Urticaria, | Angio-oedeem |
Algemene aandoeningen en toedieningsplaatsstoornissen | Reacties op de injectieplaats** |
| Griepachtige ziekte |
* Waaronder met name orofaryngeale pijn, rhinorroe, niezen
** Waaronder erytheem/roodheid, jeuken, zwelling, pijn/gevoeligheid
Beschrijving van geselecteerde bijwerkingen
Lokale reacties op de injectieplaats
Lokale reacties op de injectieplaats, waaronder erytheem/roodheid, jeuk, zwelling en pijn/gevoeligheid, werden gemeld bij 6,1% van de met alirocumab behandelde patiënten, versus 4,1% in de controlegroep (die placebo-injecties kregen). De meeste reacties op de injectieplaats waren van voorbijgaande aard en licht van ernst. Het aantal patiënten dat de behandeling staakte vanwege lokale reacties op de injectieplaats was vergelijkbaar tussen de twee groepen (0,2% in de alirocumabgroep versus 0,3% in de controlegroep). In het onderzoek naar cardiovasculaire uitkomsten (ODYSSEY OUTCOMES) kwamen reacties op de injectieplaats ook vaker voor bij patiënten die met alirocumab werden behandeld dan bij patiënten die met placebo werden behandeld (3,8% alirocumab versus 2,1% placebo).
Algemene allergische reacties
Algemene allergische reacties werden vaker in de alirocumabgroep gemeld (8,1% van de patiënten) dan in de controlegroep (7,0% van de patiënten), voornamelijk vanwege een verschil in de incidentie van pruritus. De waargenomen gevallen van pruritus waren doorgaans licht van ernst en van voorbijgaande aard. Daarnaast zijn zeldzame en soms ernstige allergische reacties, zoals overgevoeligheid, nummulair eczeem, urticaria en overgevoeligheidsvasculitis, gemeld in gecontroleerde klinische onderzoeken (zie rubriek 4.4). In het onderzoek naar cardiovasculaire uitkomsten (ODYSSEY OUTCOMES) waren algemene allergische reacties bij patiënten die met alirocumab werden behandeld vergelijkbaar met die bij patiënten die met placebo werden behandeld (7,9% alirocumab, 7,8% placebo). Er is geen verschil waargenomen in de incidentie van pruritus.
Speciale populaties
Ouderen
Bij patiënten ouder dan 75 jaar zijn geen veiligheidsproblemen waargenomen. De gegevens bij deze leeftijdsgroep zijn echter beperkt.
In de gecontroleerde fase 3-onderzoeken naar primaire hypercholesterolemie en gemengde dyslipidemie waren 1.158 (34,7%) van de met alirocumab behandelde patiënten ≥65 jaar oud en 241 (7,2%) van de met alirocumab behandelde patiënten ≥75 jaar oud. In het gecontroleerd onderzoek naar cardiovasculaire uitkomsten waren 2.505 met alirocumab behandelde patiënten (26,5%) ≥ 65 jaar oud en 493 met alirocumab behandelde patiënten (5,2%) ≥ 75 jaar oud. Er werden geen significante verschillen in veiligheid en werkzaamheid waargenomen met het toenemen van de leeftijd.
Pediatrische patiënten
De veiligheid en werkzaamheid van Praluent werden vastgesteld bij kinderen en jongeren met heterozygote familiaire hypercholesterolemie (HeFH). Een klinische studie om de effecten van Praluent te evalueren werd uitgevoerd bij 153 patiënten in de leeftijd van 8 tot 17 jaar met HeFH. Er werden geen nieuwe veiligheidsbevindingen vastgesteld en de veiligheidsgegevens bij deze populatie waren consistent met het bekende veiligheidsprofiel van het geneesmiddel bij volwassenen met HeFH.
De ervaring met alirocumab bij pediatrische patiënten met homozygote familiaire hypercholesterolemie (HoFH) is beperkt tot 18 patiënten in de leeftijdscategorie van 8 tot 17 jaar. Er zijn geen nieuwe veiligheidsgegevens waargenomen ten opzichte van het bekende veiligheidsprofiel bij volwassenen.
4 weken doseringsstudie
Het veiligheidsprofiel bij patiënten behandeld met een doseringsregime van 300 mg om de 4 weken (maandelijks) was vergelijkbaar met het veiligheidsprofiel zoals beschreven voor de klinische onderzoeksprogramma’s waarin een 2 weken doseringsregime werd gebruikt, behalve voor een hogere frequentie van lokale reacties op de injectieplaats. Lokale reacties op de injectieplaats werden in het algemeen gerapporteerd met een frequentie van 16,6% in de behandelgroep van 300 mg om de 4 weken en 7,9% in de placebogroep.
Patiënten in de alirocumab behandelgroep van 300 mg om de 4 weken kregen afwisselend placebo-injecties om de blinding te handhaven met betrekking tot de injectiefrequentie. Naast de reacties op de injectieplaats (ISR’s) die voorkwamen na deze placebo-injecties, was de frequentie van ISR’s 11,8%. Het percentage van stopzetting te wijten aan reacties op de injectieplaats was 0,7% in de 300 mg om de 4 weken behandelgroep en 0% in de placebogroep.
LDL-C-waarden <25 mg/dl (<0,65 mmol/l)
In alle klinische onderzoeken konden de lipidenverlagende achtergrondbehandelingen niet worden aangepast door de onderzoeksopzet. Het percentage patiënten dat LDL-C-waarden < 25 mg/dl (< 0,65 mmol/l) bereikte, was afhankelijk van zowel de baseline LDL-C als de dosis alirocumab.
In samengevoegde gecontroleerde onderzoeken met een aanvangsdosis van 75 mg om de 2 weken (Q2W) en een dosistoename tot 150 mg Q2W als het LDL-C van de patiënt niet < 70 mg/dl of < 100 mg/dl (1,81 mmol/l of 2,59 mmol/l) was, had 29,3% van de patiënten met een baseline LDL-C < 100 mg/dl en 5,0% van de patiënten met een baseline LDL-C ≥ 100 mg/dl en behandeld met alirocumab twee opeenvolgende LDL-C-waarden < 25 mg/dl (< 0,65 mmol/l). In het ODYSSEY OUTCOMES onderzoek, waarin de aanvangsdosis alirocumab 75 mg Q2W was en de dosis werd verhoogd tot 150 mg Q2W als het LDL-C van de patiënt niet < 50 mg/dl (1,29 mmol/l) was, had 54,8% van de patiënten met een baseline LDL-C < 100 mg/dl en 24,2% van de patiënten met een baseline LDL-C ≥ 100 mg/dl en behandeld met alirocumab twee opeenvolgende LDL-C-waarden < 25 mg/dl (< 0,65 mmol/l).
Hoewel er in de onderzoeken met alirocumab geen ongewenste gevolgen van zeer laag LDL-C werden geïdentificeerd, zijn de langetermijneffecten van aanhoudende zeer lage LDL-C-waarden onbekend.
Immunogeniciteit/‘anti-drug-antibodies’ (ADA)
In het ODYSSEY OUTCOMES onderzoek werden bij 5,5% van de met alirocumab 75 mg en/of 150 mg om de 2 weken (Q2W) behandelde patiënten antistoffen tegen het geneesmiddel (‘anti-drug-antibodies’, ADA) waargenomen na start van de behandeling, in vergelijking met 1,6% van de met placebo behandelde patiënten; de meeste hiervan waren voorbijgaande reacties. Bij 0,7% van de met alirocumab behandelde patiënten en 0,4% van de met placebo behandelde patiënten werden aanhoudende ADA-reacties waargenomen. Bij 0,5% van de met alirocumab behandelde patiënten en <0,1% van de met placebo behandelde patiënten werden reacties van neutraliserende antistoffen (NAb’s) waargenomen.
Reacties van antistoffen tegen het geneesmiddel, waaronder NAb’s, hadden een lage titer en hadden blijkbaar geen klinisch betekenisvolle invloed op de werkzaamheid of veiligheid van alirocumab, met uitzondering van een hoger percentage reacties op de injectieplaats bij patiënten met optredende ADA tijdens de behandeling in vergelijking met ADA-negatieve patiënten (7,5% t.o.v. 3,6%). De langetermijngevolgen van doorlopende behandeling met alirocumab bij aanwezigheid van ADA zijn onbekend.
In tien samengevoegde placebogecontroleerde en actiefgecontroleerde onderzoeken bij patiënten die behandeld werden met 75 mg en/of 150 mg Q2W alirocumab alsook in een apart klinisch onderzoek bij patiënten behandeld met 75 mg alirocumab Q2W of 300 mg alirocumab om de 4 weken (inclusief enkele patiënten met dosistoename tot 150 mg Q2W), was de detectie-incidentie van ADA en NAb’s vergelijkbaar met de hierboven beschreven resultaten uit het ODYSSEY OUTCOMES onderzoek.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via België: Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten: www.fagg.be – Afdeling Vigilantie: Website: www.eenbijwerkingmelden.be – E-mail: adr@fagg-afmps.be.
Nederland: Nederlands Bijwerkingen Centrum Lareb – Website: www.lareb.nl.

7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Sanofi Winthrop Industrie
82 avenue Raspail
94250 Gentilly
Frankrijk
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/15/1031/001
EU/1/15/1031/002
EU/1/15/1031/003
EU/1/15/1031/004
EU/1/15/1031/005
EU/1/15/1031/006
EU/1/15/1031/007
EU/1/15/1031/008
EU/1/15/1031/009
EU/1/15/1031/010
EU/1/15/1031/011
EU/1/15/1031/012
EU/1/15/1031/013
EU/1/15/1031/014
EU/1/15/1031/015
EU/1/15/1031/016
EU/1/15/1031/017
EU/1/15/1031/018
EU/1/15/1031/019
EU/1/15/1031/020
EU/1/15/1031/021
EU/1/15/1031/022
EU/1/15/1031/023
EU/1/15/1031/024
10. DATUM VAN HERZIENING VAN DE TEKST
18/11/2024
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau http://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3365103 | PRALUENT 75MG OPL INJ VOORGEV.PEN 2 X 75MG | C10AX14 | € 605,78 | - | Ja | - | - |
| 3365111 | PRALUENT 150MG OPL INJ VOORGEV.PEN 2 X 150MG | C10AX14 | € 605,78 | - | Ja | - | - |
| 3365129 | PRALUENT 75MG OPL INJ VOORGEV.PEN 6 X 75MG | C10AX14 | € 1438,67 | - | Ja | - | - |
| 3365137 | PRALUENT 150MG OPL INJ VOORGEV.PEN 6 X 150MG | C10AX14 | € 1438,67 | - | Ja | - | - |
| 4565875 | PRALUENT 300MG OPL INJ VOORGEVULDE PEN 3 X 300MG | € 1438,67 | - | Ja | - | - |